Can You Draw Back Residual on a Core Pack
Abstract
The tumour suppressor p53 is mutated in half of all human cancers, most frequently with missense substitutions in its core domain. We present a new assessment of the mutation database based on quantitative folding and DNA-binding studies of the isolated core domain. Our data identify five distinct mutant classes that correlate with four well-defined regions of the core domain structure. On extrapolation to 37°C the wild-type protein has a stability of 3.0 kcal/mol. This also emerges as an oncogenic threshold: all β-sandwich mutants destabilized by this amount (50% denatured) are expected to promote cancer. Other weakly destabilizing mutations are restricted to loop 3 in the DNA-binding region. Drugs that stabilize mutant p53 folding have the potential to reactivate apoptotic signalling pathways in tumour cells either by transactivation-dependent or independent pathways. Using an affinity ligand as a proof of principle we have recovered the thermodynamic stability of the hotspot G245S. With reference states for the five mutant classes as a guide, future therapeutic strategies may similarly stabilize partially structured or binding states of mutant p53 that restore limited p53 pathways to tumour suppression.
Introduction
The nuclear phosphoprotein p53 is a potent tumour suppressor, activating genes in response to cellular stress for cell cycle arrest (Kastan et al., 1991; Chan et al., 1999), or apoptosis if repair is not possible (Polyak et al., 1997). Latent function is tightly controlled by posttranslational modifications at N- and C-termini that regulate the quaternary structure of the p53 tetramer (Hupp, 1999; Jayaraman and Prives, 1999). However, 95% of tumorigenic p53 mutations are simply distributed in the central core domain where they abrogate the protein fold and its sequence-specific DNA binding affinity (Cho et al., 1994). Significantly, the mutation of one allele becomes oncogenic as a functional tetramerization domain produces a dominant negative effect (Shaulian et al., 1992; Chène, 1998; Chène and Bechter, 1999).
The crystal structure of p53 core domain bound to a consensus DNA sequence has been solved to 2.2 Å resolution (Cho et al., 1994). A large β-sandwich provides a scaffold for a conserved DNA-binding region consisting of a loop–sheet–helix motif and two large loops tethered by a zinc ion. These elements make contacts with the major and minor grooves, respectively. With a p53 tumour database of over 7500 single missense point mutations much has been learnt about the nature of mutant p53 (Hernandez-Boussard et al., 1999) (http://www.iarc.fr/p53/homepage.htm). The most frequently mutated residues fall into regions of high sequence conservation (Soussi and May, 1996; Walker et al., 1999) and either contact DNA directly or stabilize the surrounding structure (Cho et al., 1994). These hotspot sites also typically contain CpG dinucleotides or elements associated with known carcinogens (Hollstein et al., 1991; Bennett et al., 1999).
The functional status of p53 can be a marker for successful cancer therapy (Lowe et al., 1994; Kirsch and Kastan, 1998). Many inactive p53 mutants have been found to recover some level of transactivation when expressed at reduced temperatures (Zhang et al., 1994; Di Como and Prives, 1998). Thus, stabilizing the core domain with a drug specific for the native state presents one of several attractive cancer therapies aimed at restoring p53 function (Harris, 1996; Wiman, 1998; Fahraeus et al., 1999). Promisingly, p53 apoptotic responses are favoured in tumour cells with deregulated E2F-1 and can be induced by temperature-sensitive p53 at permissive temperatures (Levine, 1997; Bates and Vousden, 1999). To further this approach it is important to assess the degree of folding retained in mutant proteins at physiological temperature; wide ranging structural perturbations in the core domain may not only abrogate p53-mediated transcription, but also interfere with neighbouring sequences important for transcription-independent functions of p53 (Walker and Levine, 1996; Wang et al., 1996; Haupt et al., 1997b).
We have previously shown that the thermodynamic stability of the core domain must be measured at lower temperatures to prevent irreversible denaturation (Bullock et al., 1997). Here we present further experiments that extrapolate stability to 37°C and rationalize biochemical studies of both the core and full-length proteins. Additionally, we have completed a systematic study of 18 cancer-associated mutants that are distributed throughout the core domain and are representative of the p53 mutation database. Their phenotypes and potential for rescue have been examined firstly, by the amount of folded protein expected at 37°C, and secondly, by the relative DNA-binding affinity of the folded protein. Using these two parameters we can define, combinatorially, six possible phenotypic classes. Significantly, our data identify five mutant classes distinct from the wild type that correlate with four well-defined regions of the core domain structure. This simple pattern allows us to draw general conclusions about the p53 mutation database. These are consistent with protein engineering studies of other model proteins (Sauer and Lim, 1992; Fersht and Serrano, 1993; Munoz and Serrano, 1996), and further our general understanding of protein misfolding and disease (Thomas et al., 1995; Radford and Dobson, 1999).
Results
Folding monitored by protein fluorescence
On excitation at 280 nm, p53 core domain emits fluorescence from both tyrosines and a single tryptophan. Denaturation of p53 core domain can be monitored by changes in the fluorescence emission spectrum of either species. Measuring denaturation using a combination of the two spectral changes, fluorescence at 305 nm–fluorescence at 356 nm, provides structural probes throughout p53 core domain and increases the total signal response (the results from separate data sets at 305 nm and 356 nm are in good agreement). In the native state, tyrosine emission has high intensity with a maximum at ∼305 nm that extends to converge with a low intensity tryptophan emission maximum that is not resolved. These signals are reversed in the denatured state: the tyrosine emission intensity is decreased, while tryptophan fluorescence increases greatly with the maximum wavelength shifting to ∼350 nm (Figures 1 and 2).

Example of fluorescence spectra of wild type p53 core domain during denaturation of 10°C. Protein was buffered in 50 mM sodium phosphate pH 7.2, 5 mM DTT
Full size image

Reversible denaturation of the oncogenic mutant R249S at 10°C. Protein was buffered in 50 mM sodium phosphate pH 7.2, 5 mM DTT. The fluorescence changes at 356 nm are shown for protein initially buffered in 0M (N-state) and 5M urea (D-state) and overlay showing reversible denaturation. Denaturation monitored by fluorescence at 305 nm coincides with the data at 356 nm. Fluorescence at 317 nm is invariant supporting a two-state transition
Full size image
Fractionally greater solvent exposure is suggested for destabilized mutants relative to wild type. Thus, in the native state, mutants such as R175H and R249S in the DNA-binding region have reduced tyrosine fluorescence intensity, while V143A, close to Trp 146 in the β-sandwich, has increased tryptophan fluorescence. In agreement with NMR studies (Wong et al., 1999), the subtle differences in the native fluorescent spectra may reflect local changes in structure and dynamics in R175H and R249S, whereas the mutants G245S and R273H, which are less destabilized, have wild type fluorescent spectra.
Mutant core domain is also more prone to aggregation, which produces a highly fluorescent species characterized by a fluorescence maximum at ∼340 nm (Bullock et al., 1997). This fluorescence increases in intensity proportionally to the degree of aggregation, for example during thermal denaturation. The conformational status of p53 core domain at different pH values is also reported by protein fluorescence. These spectral changes coincide precisely with changes in fluorescence on binding ANS (8-anilinonaphthalene-1-sulphonic acid) and define a pH denaturation curve with maximum stability at pH 7.2. We have previously reported an apparent T m of 42°C, while at pH 6.0 this falls to 36°C (Bullock et al., 1997). Below pH 5.5 large increases in tryptophan and ANS fluorescence show a transition to an acid molten globule state.
Thermodynamic stability of wild type and the six p53 hotspots in cancer
The structure of p53 core domain consists of two elements: a β-sandwich with an immunoglobulin superfamily fold and a conserved DNA-binding region consisting of zinc co-ordinated loops 2 and 3 and a loop–sheet–helix motif (Cho et al., 1994). The β-region is comparatively hydrophobic (estimated pI=6.3, MacVector™ 6.5, Oxford Molecular Group) and is mostly an infrequent target for mutation. In contrast, the DNA-binding region is basic as expected (estimated pI=10.4) and forms the site of the most frequent cancer mutations. The thermodynamic stability and DNA-binding characteristics of tumorigenic p53 mutants can also be divided into classes corresponding to their location within these regions of substructure (Figure 4).

Analysis of residual folding and DNA-binding activity in p53 core domain mutants at 37°C. (a) DNA contact, Zn2+ region and β-sandwich mutants fall into well-defined classes with common phenotypes. Other mutations scattered throughout the DNA-binding region are more varied. Heavily destabilized mutants in the Zn2+ region and β-sandwich are coloured pink and red, respectively, while DNA contact and surrounding residues, which can be less destabilized, are coloured dark and light blue, respectively. A free energy of unfolding, in water 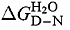 , of 0 kcal/mol (shown by a feint dashed line) corresponds to 50% denatured protein. (b) Ribbon representation of p53 core domain. The DNA-binding region and β-sandwich are coloured green and yellow, respectively. Tumorigenic mutations studied are represented by spacefilled atoms and coloured according to a. The bound Zn2+ ion is displayed as a white spacefilled atom. Figure prepared with the program MOLMOL (Koradi et al., 1996)
, of 0 kcal/mol (shown by a feint dashed line) corresponds to 50% denatured protein. (b) Ribbon representation of p53 core domain. The DNA-binding region and β-sandwich are coloured green and yellow, respectively. Tumorigenic mutations studied are represented by spacefilled atoms and coloured according to a. The bound Zn2+ ion is displayed as a white spacefilled atom. Figure prepared with the program MOLMOL (Koradi et al., 1996)
Full size image
At 25°C, wild type p53 core domain has a free energy of unfolding, of 7.5 kcal/mol (Table 2), which falls within the normal range for single domain proteins (5–15 kcal/mol). However, the stability decreases sharply with increasing temperature leading to irreversible denaturation with an apparent T m=42°C and an increased tendency to aggregate (Bullock et al., 1997). All tumorigenic mutants studied here are destabilized. The difference in free energy between wild type and mutant p53 core domain, 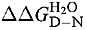 , is, therefore, measured at 10°C to avoid aggregation. Wild type has a free energy of unfolding of 10.2 kcal/mol at this temperature (Table 1).
, is, therefore, measured at 10°C to avoid aggregation. Wild type has a free energy of unfolding of 10.2 kcal/mol at this temperature (Table 1).
Full size table
Full size table
Six residues in p53 have been identified as hotspots for tumorigenic mutations. All six occur within the DNA-binding substructure. In this region, mutations can remove DNA contacts, or have a structural effect, destabilizing the local conformation or inducing global denaturation. The four 'structural' hotspots R175H, G245S, R249S and R282W have one predominant substitution while the two 'DNA-contact' hotspots, Arg 248 and Arg 273, both have two: R248Q, R248W, R273C and R273H. For the latter class the most common mutation has been made according to the January 1999 IARC p53 mutation database. The five arginine hotspot residues are distinguished as the only side chain groups in this study to form hydrogen bond interactions in the wild type structure. With the significant exception of Arg 175, these side chains are also among the most solvent exposed groups studied (Table 3).
Full size table
We have previously reported the free energy of unfolding of four hotspot mutations including R175H, R248Q, R249S and R273H (Bullock et al., 1997). We have since found that overnight incubation in urea at 10°C is preferable to achieve complete equilibrium (see Materials and methods) and have repeated these measurements and extended them to include many other mutants including G245S and R282W. The tryptophan introduced at position 282 fluoresces strongly without change upon denaturation suggesting full solvent exposure in the folded state. This masks much of the signal from Trp 146, the main probe for denaturation, but the standard analysis is still possible due to the additional changes in tyrosine fluorescence.
Tumorigenic mutations at the two key DNA-contact residues, Arg 248 and Arg 273, prevent DNA-binding by the isolated core domain, although other contacts may enable full-length p53 to bind weakly (without transactivation activity) (Bargettoni et al., 1993). R273H has similar stability to wild type, =0.5 kcal/mol (Table 1). R248Q in loop 3, however, appears similar to the structural mutant R249S (see below); with no DNA-binding activity and a 1.9 kcal/mol loss in stability. This similarity has also been observed by NMR (Wong et al., 1999). Some structural distortion may occur through the main chain hydrogen bond between Arg 248 and Ser 240, which lies between the two zinc ligands, Cys 238 and Cys 242.
At the protein exterior, the conformation of loop 3 is sensitive to mutations with modest destabilization energies, as exemplified by the hotspots, G245S and R249S (=1.2 and 1.9 kcal/mol, respectively). Gly 245 facilitates a tight turn within loop 3 and is the only non-arginine cancer hotspot. The mutant G245S retains 63% of the wild type affinity for the gadd45 promoter at 20°C, but its activity may be compromised at 37°C (Table 1). This phenotype provides the most amenable target for drug-based rescue. As a proof of principle we have been able to recover its thermodynamic stability using the affinity ligand heparin (Table 1). Arg 249 forms hydrogen bonds with the main chains of Gly 245 and Met 246 (L3 loop) and a salt bridge with Glu 171 (loop L2) (Cho et al., 1994). Its hotspot mutant, R249S, has lost all DNA-binding activity (Table 1). NMR studies have revealed local changes in the structure and dynamics of this mutated region at 20°C that reduce the complementarity of p53 core domain for the DNA surface (Wong et al., 1999).
Arg 175, within loop 2, is buried in a polar region between loops 2 and 3 and precedes the zinc ligand Cys 176. In addition to several important hydrogen bonds, it also forms a salt bridge with Asp 184 in the 53BP2 (Gorina and Pavletich, 1996) and non-consensus DNA complexes (Cho et al., 1994). R175H, the most common structural mutant, is significantly destabilized, =3.5 kcal/mol, and fails to bind DNA. This mutation shows a common property of p53 cancer mutations: the destabilizing effects of newly introduced contacts may be of greater significance than those resulting from the removal of previously stabilizing interactions. Alternative substitutions at Arg 175 produce many different mutant phenotypes (Ryan and Vousden, 1998). To test the hypothesis that this reflects a variation in the folding of the mutant we have analysed the mutant R175A, which does not occur in cancer, for stability and DNA-binding affinity. A large difference in stability between R175H and R175A is indeed found. The latter has only a small loss in stability, =0.7 kcal/mol, and marginally reduced DNA-binding affinity at 20°C (67% of wild type). Unlike R175H, the lost contacts in R175A may be partially compensated for by the close proximity of Arg 196, which can also interact with Asp 184.
The final structural hotspot, Arg 282, is located in the DNA-binding helix (H2) and packs between this and the surface of β-strands S2 and S2′. Substitution of a trytophan moiety has a large thermodynamic cost, =3.3 kcal/mol. This side chain is too large to bury without severe disruption of structure and from the fluorescence appears to create a solvent-accessible environment. When purified and studied at 20°C as the isolated core domain, R282W retains 82% of the wild type DNA-binding affinity (Table 3). This may suggest that the DNA contacts Arg 248 and Arg 273 (closer to Arg 249) provide more binding energy than Arg 280 and Lys 120 (closer to Arg 283). At 37°C, R175H and R282W are expected to be sufficiently destabilizing to cause denaturation (see below).
Other mutations in the DNA-binding region
Tumorigenic mutations at the zinc-binding site (residues Cys 176, His 179, Cys 238 and Cys 242) occur as minor hotspots in comparison with R175H, which is the most common structural p53 mutation in cancer. Three mutations, R175H (loop 2), M237I (loop 3) and C242S (loop 3), were made in this region. Cys 242 binds zinc directly, while Arg 175, adjacent to the ligand Cys 176, makes several stabilizing hydrogen bonds to loops 2 and 3, including two to the backbone carbonyl of Met 237, which neighbours the ligand Cys 238. We have previously shown using a spectrophotometric assay that C242S expressed at 20°C remains bound to zinc during urea denaturation with 1 : 1 stoichiometry (Bullock et al., 1997). In fact, with a total of ten cysteines and nine histidines, the core domain readily accepts metal ions: the addition of excess zinc causes aggregation.
The stability of wild type p53 core domain in the presence of 10 mM EDTA is reduced by 3.0 kcal/mol and the protein has no detectable DNA-binding activity. These characteristics are also observed for the mutated zinc ligand C242S (=3.1 kcal/mol) and EDTA treatment of this mutant has little effect (Table 1). (By comparison, the more distant hotspot R249S, loses an additional 0.9 kcal/mol in 10 mM EDTA). In addition to the critical role of the cysteine side chain, the backbone oxygen of Cys 242 is involved in hydrogen bonding with the backbone amides of four loop 3 residues, Gly 244, Gly 245, Met 246 and Asn 247, and helps to present Arg 248 for its minor groove contacts. M237I (loop 3) appears similar to the hotspot mutant R175H (loop 2), with a 3.2 kcal/mol loss in stability and less than ∼15% of the wild type DNA-binding affinity. Both mutations involve side chain truncations, but introduce bulkier groups. Increased solvent accessibility is suggested by changes in fluorescence and proteolytic degradation (see Materials and methods). These results are consistent with antibody studies which show that both mutants bind to PAb240, but not to PAb1620 (Ory et al., 1994; Rolley et al., 1995).
F134L lies in β-strand S2′, which is part of the three-stranded sheet in the DNA-binding loop–sheet–helix motif. In the wild type structure, the aromatic ring of Phe 134 makes a van der Waals contact with the aliphatic portion of the hotspot Arg 282, and constitutes part of the packing between β-sheet and DNA-binding helix. Mutation to leucine will create a large cavity in this region. In comparison to the loop 3 hotspots G245S and R249S, the thermodynamic cost is therefore much greater, =4.8 kcal/mol. At 20°C the DNA-binding affinity of F134L is just 34% of wild type.
Mutations in the β-sandwich
In contrast to the DNA-binding surface, tumorigenic mutations in the β-sandwich affect hydrophobic residues in the protein core and occur with less frequency. Nine mutations have been made in this region: V143A (S3), L145Q (S3), P151S (S3-S4 loop), V157F (S4), I195T (S5), Y220C (S7-S8 loop), I232T (S8), I255F (S9), and F270C (S10). Each mutation is severely destabilizing ( ranges from 3.0–4.5 kcal/mol, Table 1), but at 20°C, all retain a DNA-binding affinity between 36 and 72% of wild type. Pro 151, Val 157 and Tyr 220 are furthest from the DNA, yet represent significant mutational hostpots within the β-sheet. Cho et al. (1994) have suggested that these residues may be important for the stability of the loop regions at this side of the β-sandwich. We can detect a slight increased tendency for these mutants (and F134L) to aggregate in vitro under denaturing conditions. However, they are not significantly distinguished from the remaining substitutions (Table 1) as no obvious relationship between distance from the DNA and stability or DNA-binding activity is apparent. The residual activity of these mutants at low temperatures supports a denatured state in tumour cells. EDTA-treatment of these mutants has an additive effect that is too destabilizing to quantify.
Extrapolating the effects of mutation to 37°C
At temperatures close to its T m, p53 core domain aggregates irreversibly. The observed T m thus depends on the concentration and the rates of heating, denaturation and aggregation of the particular protein, so melting temperatures obtained by differential scanning calorimetry (DSC) or spectroscopy are only a qualitative guide to stability. The irreversibility also prevents direct measurement of the unfolding equilibrium at 37°C. To circumvent this problem, the temperature-dependence of wild type stability has been determined by urea denaturation performed at 10, 15, 20 and 25°C (Table 2). The free energy of unfolding for wild type was then extrapolated from these data to 37°C using (4) and (5) (see Materials and methods). With knowledge of the stability of p53 core domain under physiological conditions, the degree of folding in tumorigenic mutants can be assessed from their known differences in free energy at 10°C. These calculations allow us to estimate the stability and fraction folded of both wild type and mutant p53 core domain over the relevant temperature range (Figure 3a,b).

Extrapolated temperature denaturation curves for p53 core domain. (a) The temperature dependence of p53 core domain stability was calculated from data in Table 2 using the Gibbs-Helmholtz (5) and plotted for wild type and hypothetical mutants destabilized by 2 and 4 kcal/mol. A theoretical value ΔC p=3.8 kcal/(mol K) was calculated from the estimated change in solvent accessible surface area in p53 core domain upon denaturation according to the correlation derived by Myers et al. (1995) (equation 4). Data were fitted by non-linear least squares analysis using the Kaleidagraph ™ program (Abelbeck Software). For wild type: ΔG 37°C=3.0 kcal/mol; ΔH Tm=169 kcal/mol; T m=316 K (43°C). (b) Unfolding transitions for wild type and hypothetical mutants destabilized by 2 and 4 kcal/mol were calculated from (a) using the standard equation ΔG=−RTIn([D]/[N]). The feint dashed line at 37°C illustrates that a relatively small change in the fraction unfolded occurs upon 2 kcal/mol destabilization, while a large change is seen between 2 and 4 kcal/mol
Full size image
The melting temperatures (where ΔG T=0) for wild type, R249S and V143A are estimated at 43, 39 and 36°C, respectively. At 37°C their expected free energies of unfolding are 3.0 kcal/mol (99% folded) 1.1 kcal/mol (85% folded) and −0.5 kcal/mol (31% folded). Table 1 reports the calculated fraction folded at 37°C for all tumorigenic mutants studied here. It is clear from these results that most mutants of p53 core domain are at least 50% denatured at physiological temperature. Less destabilized mutants can be found in the DNA-binding region in loop 3. The apparent melting temperature for wild type has been calculated by differential scanning calorimetry to be 42°C (Bullock et al., 1997). The actual melting temperature is expected to be slightly higher than this as irreversible aggregation during measurement leads to a steeper unfolding transition. These estimates seem, therefore, to be of sufficient quality to make useful conclusions about p53 core domain folding.
Discussion
Tumorigenic mutations destabilize p53 core domain
The abrogation of p53 activity due to destabilizing mutations is thought to be a key event in tumour development. The crystal structure of p53 core domain has provided a framework for understanding the role of these tumorigenic mutations in cancer. Its structure can be divided into a β-sandwich scaffold and its supported DNA-binding region that contains a loop–sheet–helix motif and two loops co-ordinating a single Zn2+ ion. We have previously reported an assay for the thermodynamic stability of p53 core domain that allows the effects of mutation to be measured. Using this assay we have completed a survey of 18 cancer-associated mutations distributed throughout the domain. Of these, the structural mutations destabilize the protein by between 1.2 and 4.8 kcal/mol.
In recent years empirical values for the destabilizing effects of various mutations have been established using model protein systems (Fersht, 1998). One of the most comprehensive studies used the ribonuclease barnase to examine the effects of removing hydrogen bond partners and hydrophobic side chains (Serrano et al., 1992). Isolated charged residues caused a 3–4 kcal/mol loss in stability in buried positions, but only 0.3–1 kcal/mol at the protein surface. Similarly, removal of the hydrogen bond partner of an uncharged group reduced stability by 0.5–2 kcal/mol, depending on solvent exposure. Side chain truncations were found to cost an average of 1.5 kcal/mol per methyl(ene) group deleted when buried, or 0.8 kcal/mol close to the protein surface.
Tumorigenic mutations in p53 core domain typically involve a combination of detrimental effects, but are in agreement with the values above. Some conservative mutations (e.g. V143A) are directly comparable, while others (e.g. I232T) involve additive effects. The results for the hotspot arginine mutations R175H and R249S (=3.5 and 1.9 kcal/mol) also reflect their relative solvent exposures (Tables 1 and 3). The individual interactions broken and gained by non-conservative mutations, such as V157F, F270C and R282W, cannot be easily interpreted. However, the large number of neighbouring side chains that are affected dictates that these substitutions are strongly destabilizing (Tables 1 and 3). Cancer has selected many mutations of this type in p53.
Our aim in presenting quantitative values for these mutations is to provide a high resolution understanding of the mutant p53 phenotype that will further refine the 'antibody' view of mutant folding (Milner, 1995). In combination with cellular assays for the full-length protein, these results provide an insight into the p53 mutation database as a whole. We hope these definitions will serve as a valuable resource and reference point for future cellular and therapeutic studies.
Defining the effects of tumorigenic mutations in p53 core domain
The range of thermodynamic stabilities can be understood by their probable affect on the folded state at 37°C. Native structure is retained in some DNA-contact mutants where there is no loss in stability ( ⩽0.5 kcal/mol). Weakly ( ⩽2.0 kcal/mol) and strongly ( ⩾3.0 kcal/mol) destabilizing mutations will cause disruption of local structure and global denaturation, respectively. When further considering the DNA-binding activities of these mutants at permissive temperatures for folding (e.g. 20°C), we can define a wild type and five mutant p53 classes (Table 4). Using these definitions, four 'determinant' structural regions are identified: DNA contact, DNA-binding region, zinc-binding region and β-sandwich (Figure 4a,b). The mutant phenotypes that become oncogenic are highly specific in all but the DNA-binding region, where all mutations are potentially damaging.
Full size table
Nearly 40% of missense mutations in the core domain occur at arginine residues; the next two most frequently mutated residues, glycine and cysteine, are each five times less frequent. The effects of mutation at these last two residues are easily understood: substitutions for glycine increase the side chain length and perturb conformation (e.g. G245S), while cysteine residues play a key role in zinc ligation (e.g. C242S). A dichotomy is observed between arginine residues in the DNA-binding region and the β-sandwich. In the former region hotspot arginines either form DNA contacts or bury their guanidinium side chains for van der Waals, hydrogen bond and salt bridge interactions (Cho et al., 1994). In contrast, arginines in the β-sandwich are solvent exposed and likely to yield weakly destabilizing mutations. However, more importantly, four arginine hotspots contain CpG dinucleotides (Arg 174, 248, 273 and 282) and the fifth (Arg 249) is associated with exposure to aflatoxin B1 (Hollstein et al., 1991). This mutability is compounded by substitution to histidine and tryptophan, which introduce destabilizing steric clashes.
Correlation with observed p53 behaviour in vivo
The cellular half-life of wild type p53 is kept low by negative regulatory feedback involving Mdm2 (Haupt et al., 1997a). Inactive mutant p53 is unable to stimulate transcription and, in the absence of Mdm2, mutant p53 protein accumulates (Midgley and Lane, 1997). Thus, cellular half-life is not correlated to p53's thermodynamic stability. However, the destabilization of mutant p53 is evident from its association with the HSP70 class of chaperone (Pinhasi-Kimhi et al., 1986; Hinds et al., 1990; Fourie et al., 1997).
Direct study of p53 folding and stability at 37°C is difficult as the kinetics of p53 unfolding and aggregation may be significant. Nevertheless, the low thermostability of full-length tetrameric p53 has been well documented (Friedlander et al., 1996; Hansen et al., 1996; Ponchel and Milner, 1998). This protein loses DNA-binding activity after 5 min at 42°C, but not at 40°C (Hansen et al., 1996). However, even at 37°C, irreversible denaturation and aggregation (beyond tetramer) is sufficient to cause complete loss of DNA binding and the PAb1620(+) conformation within 1 h (Hansen et al., 1996). Interestingly, Ponchel and Milner (1998) have found higher levels of p53 transactivation in vivo at 32°C than 37°C. The apparent melting temperature of the isolated core domain appears unchanged, apparent T m=42°C (Bullock et al., 1997). Importantly, the results for the core domain are in excellent agreement with the antibody-binding studies using PAb1620 and PAb240, raised against native and denatured p53, respectively (Ory et al., 1994; Rolley et al., 1995).
We have measured the temperature dependence of p53 core domain stability between 10 and 25°C, where there is no aggregation, and used this to extrapolate to 37°C (Figure 3a,b). At this temperature the native wild type structure is estimated to be 3.0 kcal/mol more stable than its denatured state (Table 2). Mutants of p53 core domain destabilized by 3.0 kcal/mol are therefore expected to be 50% denatured. Some mutants, such as R249S, which does not bind DNA, are less destabilized; 85% of these molecules may be folded. In vivo studies show that R175A has 77% of the wild type transactivation level (Thukral et al., 1994), whereas the cancer substitution, R175H, has no activity (Ory et al., 1994). Similarly, in the isolated core domain R175A has 67% of the wild type DNA-binding affinity at 20°C, while R175H does not bind (Table 1). We have found a significant difference between the thermodynamic stability of the two core domain mutants: R175H, =3.5 kcal/mol (est. 30% folded at 37°C) and R175A, =0.7 kcal/mol (est. 98% folded at 37°C). Further substitutions at Arg 175 have yielded proteins with either full wild type activity (e.g. R175C) or with cell cycle arrest, but not apoptotic function (e.g. R175K) (Ryan and Vousden, 1998). Again, these differences appear to reflect alterations in the folding of the core domain at 37°C: all of the Arg 175 mutants showing some transactivation have been shown to be PAb1620(+)/PAb240(−)/Hsp70(−), while those with no activity (e.g. R175F) were PAb1620(−)/PAb240(+)/Hsp70(+) (Ory et al., 1994). These classes are consistent with the destabilizing effects of larger side chain substitutions.
The β-sandwich mutant V143A is temperature sensitive for transactivation in vivo (Zhang et al., 1994). At 20°C, this core domain mutant has 68% of the wild type DNA-binding affinity, but it is oncogenic at 37°C because it is denatured (Table 1). Similar sub-physiological activity has been shown for β-sandwich mutants, including V143A and Y220C, in a yeast-based assay for transactivation (Di Como and Prives, 1998). Together these studies suggest that the recovery is biased towards promoters specific for cell cycle arrest rather than apoptotic function (Zhang et al., 1994; Di Como and Prives, 1998; Ryan and Vousden, 1998). Extrapolation of V143A stability to the permissive temperature of 32.5°C suggests that its free energy of unfolding will be 1.4 kcal/mol (est. 91% folded). The wild type stability at 37°C is 3.0 kcal/mol. The incomplete recovery of V143A, relative to wild type, may be due in part to this difference and in part to its reduced DNA-binding affinity. NMR studies indicate that the reduced affinity is due to structural repacking throughout the core domain to compensate for the two deleted methyl groups (Wong et al., 1999).
Some 95% of p53 mutations occur in the core domain, but those that induce denaturation and aggregation are known to adversely affect the function of other p53 domains (Raycroft et al., 1991). Our data describing core domain folding should explain which mutants have this effect. Bimolecular collisions leading to aggregation in mutant p53 core domain will become unimolecular in the context of the full-length tetramer. Furthermore, the additional presence of other less ordered domains increases the aggregation tendency. For strongly destabilized mutants these effects are highly detrimental to both p53 function and its study using sensitive biophysical procedures, and therefore, dictate that these experiments are performed on the isolated core domain. Weakly destabilized, but folded mutant p53 core domain may retain some limited function involving protein–protein interactions. For example, residues 61–94, which constitute an SH3 domain binding motif adjacent to the core domain (Walker and Levine, 1996), may be differentially affected in this manner. Indeed, additional phenotypes have been observed in some damaged cell-lines where p53-induced apoptosis has been shown to be possible in the absence of p53-transactivation (Caelles et al., 1994; Haupt et al., 1995; Guillouf et al., 1998; Bates and Vousden, 1999). Further, a new potential p53 signalling pathway involving forkhead-associated (FHA) domains has recently been reported (Durocher et al., 1999; Yaffe and Cantley, 1999). We must wait to see whether these properties are distinguished by the definition of five mutant p53 classes.
Interpreting the p53 mutation database
The January 1999 release of the IARC p53 mutation database contains over 10 000 entries (http://www.iarc.fr/p53/homepage.htm), of which 75% are missense point mutations. The critical role of the core domain is reflected in the database, with 95% of mutations falling into this region. Rather than affecting just a few key functional residues, tumorigenic p53 mutations occur throughout the core domain structure (Figure 4a,b). Thus, p53 provides us with the best opportunity to understand the effect of disease-causing mutations on protein structure and function. Interpretation of this database should yield general lessons that can be applied to other proteins in disease (for example, the PKD domains of polycystin-1; Bycroft et al., 1999).
The mutational frequencies of individual residues are a complex function of genetic and phenotypic factors (Wacey et al., 1999). Theoretically, once a mutation is sufficiently damaging to be oncogenic, there may be little selection pressure to increase the frequency of the most destabilizing mutations. R175H and G245S have similar mutation frequencies (both result from changes at CpG dinucleotides), but in the core domain differ in stability by 2.3 kcal/mol. These residues occur in loops 2 and 3, respectively, where minor perturbations are significant due to the lack of regular secondary structure (Cho et al., 1994). Susbstantial mutation preferences do, however, distinguish between phenotypically wild type and oncogenic mutants, for example R175C and R175H have ten and 518 database entries, respectively. These substitutions represent the common C→T and G→A mutations at CpG dinucleotides. A similar bias is present at Arg 282 with tryptophan occurring in great excess over glutamine (Walker et al., 1999). However, no bias exists at the DNA-contact residues, Arg 248 and Arg 273, where either point mutation is damaging. A CpG dinucleotide linked with tobacco smoke exposure (Bennett et al., 1999) also accounts for the high occurrence of the β-sandwich mutant V157F.
Although this study has rigorously defined the effect of individual mutations, a picture of the mutation database as a whole has also emerged, with the definition of five mutant classes (Table 4). Mutations at the DNA-binding surface occur in all five categories, while only Class IV mutations are oncogenic in the β-sandwich (Table 4). At 37°C, a loss of 3.0 kcal/mol is estimated to cause 50% denaturation. This appears to represent a threshold value and explains the common effect of β-sandwich mutations (=3.0–4.5 kcal/mol) relative to structural mutations elsewhere (=1.2–4.8 kcal/mol). Mutations of this severity will be oncogenic if they occur anywhere in the core domain structure, but will be predominantly found in the hydrophobic core. Substitutions of valine for isoleucine, which can destabilize proteins by 1.5 kcal/mol, are not expected to be oncogenic in this region and form only 0.001% of the database. In total, 28% of missense mutations occur in the β-sandwich. The wider spectrum of disruptive mutations in the DNA-binding surface seems proportional to their higher occurrence in the database.
Identifying the most realistic targets for drug rescue of mutant folding
Mutations in p53 are present in half of all human cancers (Hollstein et al., 1991). Given its important role as an anti-cancer agent, many different strategies for restoring p53 tumour suppressor function to cancer cells have been suggested (Harris, 1996; Wiman, 1998; Fahraeus et al., 1999). One possible approach is to use small molecules that bind to p53 core domain to repair the effect of missense mutation, either by restoring DNA contacts, or by stabilizing the wild type conformation. However, each mutant has its own local structural changes that require correction. Furthermore, many bulky substitutions create new disruptive interactions that cannot be removed. These inherent problems are avoided by considering a generic molecule that binds to the β-sandwich: the law of mass action ensures that any ligand preferentially binding to the native state over the denatured state will increase the protein's thermodynamic stability. Many common p53 mutants in this study retain some DNA-binding affinity at low temperatures, so transcriptional activity may be recovered further by increasing their thermodynamic stability (Randell Brown et al., 1997).
Although the results using the C-terminal derived p53 peptide are encouraging (Selivanova et al., 1999), a powerful drug needs to show a stronger and specific interaction with the core domain. Our DNA-binding data at 20°C assess how far folded mutant structures are distorted from the wild type conformation. One function of a drug is to extrapolate this activity to 37°C. We have been able to restore wild type stability to the hotspot mutant G245S using the affinity ligand heparin (Table 1). In principle, this shows that drug rescue of mutant p53 is a viable therapeutic strategy to aid future anti-cancer regimes. This mutant is by far the most promising for clinical therapy as it has a high frequency in cancer and yet only small reductions in stability and DNA-binding affinity. With severe defects in both areas a far worse prognosis is apparent for mutations surrounding the zinc-binding site; this mutant class may require gene therapy for functional rescue.
Success may be more forthcoming if the requirement for core domain folding is simply to prevent aggregation with adjacent domains necessary for protein–protein interactions. In response to chemotoxic and radiotherapy, these signalling pathways may have the potential to induce p53 transcription-independent apoptosis in tumour cells, particularly those with deregulated E2F-1 (Caelles et al., 1994; Guillouf et al., 1998; Bates and Vousden, 1999). Inspection of the molecular surface suggests that potential drug-binding sites do exist in p53 core domain. With reference states for five mutant p53 classes our assay for folding and stability can now be used to monitor the progress of future candidate molecules.
The results of this study provide a quantitative basis for interpreting the mutation database and past and future experimental research on the mutant p53 protein. These studies have important implications for the assessment of disease prognoses and the relative merits of different therapeutic strategies for p53 rescue.
Materials and methods
Protein expression and purification
Human p53 core domain (residues 94–312) was subcloned into the vector pRSET(A) (Invitrogen) as described previously (Bullock et al., 1997). Protein expression and purification strategies have been optimized to allow preparation of many heavily destabilized mutants. Protein was expressed at 20°C in Escherichia coli strain BL21(DE3), or when necessary its derivative C41 which can carry a higher proportion of soluble protein (Miroux and Walker, 1996). Cultures were initially grown at 37°C to A 600=1.5 before overnight induction with 1 mM IPTG in a refrigerated shaker at 20°C. Cells were harvested by centrifugation and lysed in 50 mM Tris pH 7.2, 5 mM DTT, 1 mM PMSF using either sonication or an EmulsiFlex™-C5 high pressure homogenizer (Avestin Inc.). Soluble lysate was loaded onto a SP Sepharose cation exchange column (Pharmacia) and eluted with a NaCl gradient (100–300 mM). Further purification was achieved by affinity chromatography using a HiTrap Heparin Sepharose column (Pharmacia) in 50 mM Tris pH 7.2, 5 mM DTT with a NaCl gradient (100–300 mM). Columns were kept at 5°C using a cooling water jacket. Protein concentration was measured spectrophotometrically by the method of Gill and von Hippel (1989) after overnight dialysis against 50 mM sodium phosphate pH 7.2, 5 mM DTT. Protein was frozen in liquid nitrogen and stored in 1–2 mL aliquots at −80°C.
Several purified mutants (e.g. R175H and M237I) contain shorter proteolytic fragments by SDS–PAGE. This was also apparent from an increase in fluorescence at ∼340 nm, consistent with the presence of misfolded species (Bullock et al., 1997). These degradation products were removed using a ceramic hydroxyapatite column (Bio-Rad) with a sodium phosphate gradient (25–150 mM) and finally by gel filtration chromatography (50 mM sodium phosphate pH 7.2, 150 mM NaCl, 5 mM DTT). R175H and M237I also have decreased sensitivity to urea denaturation (low 'm' values, see below and Table 1) indicating a smaller change in surface area on denaturation. This may result from the denatured state being more compact than that of wild type or the native state being more solvent exposed (Fersht, 1998). Wild type p53 core domain also has a low m value after overnight incubation and denaturation in 0.1 mM DTT, suggesting that this effect may be due in part to the oxidation of some of the seven free cysteines.
Equilibrium denaturation
Denaturation was monitored by fluorescence using an Aminco-Bowman Series 2 luminescence spectrofluorimeter with excitation at 280 nm (bandpass 4 nm) and this emission scanned from 300–370 nm (bandpass 4 nm). Stock urea solutions were prepared volumetrically. 100 μL of protein (∼18 μM) in the relevant buffer were added to 800 μL of buffer containing the appropriate concentration of urea. Each sample was incubated at 10°C overnight for ∼14 h before fluorescence was measured using a 1 mL cuvette in a thermostatted holder. Temperature was monitored by a thermocouple in the cell. Equilibration appears close to completion at 5–6 h (as measured previously (Bullock et al., 1997)), but more consistent results are seen after a longer incubation time. This may be a consequence of the presence of 18 prolines in p53 core domain (a.a. 94–312) which equilibrate slowly between cis and trans conformations. Longer incubation times (>24 h) did not alter the results and the free energies determined using urea and the more stable guanidinium hydrochloride were in excellent agreement (Table 1).
The data were fitted to equation (1) which assumes a two-state model in which the fluorescence of the folded and unfolded states are dependent on denaturant concentration (Clarke and Fersht, 1993):
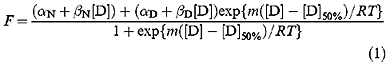
F is the fluorescence at the given [denaturant]. Previously tryptophan fluorescence at 356 nm was normalized using invariant data at ∼320 nm (Bullock et al., 1997). In this study we have not used this isofluorescent point, but instead plotted F as fluorescence at 305 nm–fluorescence at 356 nm to capture both tyrosine and tryptophan emission changes. αN and αD are the intercepts and βN and βD are the slopes of the baselines at low (N) and high (D) denaturant concentrations, respectively, [D] is the concentration of denaturant, [D]50% is the concentration of denaturant at which half of the protein is denatured, m is the slope of the transition, R is the gas constant and T is the temperature in K. The data were fitted to this equation by non-linear least squares analysis using the general curve fit option of the Kaleidagraph ™ program (Abelbeck Software), which gives the calculated values for individual experimental measurements of m and [D]50% together with their standard errors.
The free energy of denaturation of proteins in the presence of denaturant 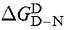 is, to a first approximation, linearly related to the concentration of denaturant (Pace, 1986) (equation 2):
is, to a first approximation, linearly related to the concentration of denaturant (Pace, 1986) (equation 2):
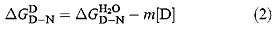
The difference in free energy of denaturation between wild type and mutant proteins may be calculated from:
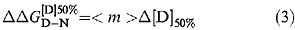
where Δ[D]50% is the difference between the value of [D]50% for wild type and mutant and <m> is the average value of m (Serrano et al., 1992).
Extrapolation of stability to 37°C
The change in solvent accessible surface area (ΔASA) upon denaturation for p53 core domain (chain A; Cho et al., 1994) is estimated to be 21 150 Å2 using the program NACCESS (Hubbard and Thornton, 1993) and an Ala-X-Ala peptide model for the denatured state (Hubbard et al., 1991). The heat capacity change for the core domain (ΔC p=3.8 kcal/mol K) was calculated from this value using the strong correlation (R=0.97) derived in equation (4) (Myers et al., 1995). A similar correlation shows excellent agreement between the predicted value of m at 25°C (−2.70 kcal/molM −1) and the experimental value (−2.79 kcal/molM −1) (Table 2). The free energy of unfolding of wild type p53 core domain measured at 10, 15, 20 and 25°C by urea denaturation was fitted to equation (5) with ΔC p fixed at 3.8 kcal/mol K. A short extrapolation of this curve to 37°C allowed core domain stability to be assessed at physiological temperature. Mutant stabilities were then estimated by subtracting each mutant's value from wild type stability (ΔG T). Values of change little with temperature over the relevant range (Dalby et al., 1998) and any error in the value of ΔC p will not significantly affect extrapolation to 37°C.
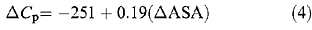
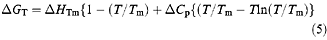
The fraction of p53 molecules folded at 37°C was estimated from ΔG T using the standard equation ΔG=−RTln([D]/[N]).
DNA binding
The binding of p53 core domain to the gadd45 promoter (5′-biotin-GTA CAG AAC ATG TCT AAG CAT GCT GGG GAC-3′ and 3′-CAT GTC TTG TAC AGA TTC GTA CGA CCC CTG-5′) was assessed at 20°C by surface plasmon resonance as described previously (Nikolova et al., 1998). Double-stranded gadd45 oligonucleotide was immobilized onto flow cells 1–3 on a streptavidin coated chip (SA) using a BIAcore™ 2000, Pharmacia Biosensor AB (Uppsala, Sweden). Flow cell 4 was left empty to determine the background response. 50 μL protein at concentrations ranging from 0.03–1 μM were injected at a flow rate of 10 μL/min over flow cells 1–4. Protein was buffered in 50 mM Tris, pH 7.2, 5 mM DTT. The change in resonance units upon binding was measured 500 s after injection and the background response subtracted. To regenerate the surface after each sample, 10 μL of 1M NaCl in 50 mM Tris, pH 7.2, 5 mM DTT were injected and proved sufficient to dissociate the p53-DNA complex. The relative response units upon binding were plotted against the logarithm of the protein concentration and the data fitted to a two-state equation (1) using the general curve fit option of the Kaleidagraph ™ program (Abelbeck Software). Relative binding affinities were derived from the transition midpoint.
References
-
Bargettoni J, Manfredi JJ, Chen XB, Marshak DR and Prives C . 1993 Genes Dev 7: 2565–2574
-
Bates S and Vousden KH . 1999 Cell Mol Life Sci 55: 28–37
-
Bennett WP, Hussain SP, Vahakangas KH, Khan MA, Shields PG and Harris CC . 1999 J Pathol 187: 8–18
-
Bullock AN, Henckel J, DeDecker BS, Johnson CM, Nikolova PV, Proctor, MR, Lane DP and Fersht AR . 1997 Proc Natl Acad Sci USA 94: 14338–14342
-
Bycroft M, Bateman A, Clarke J, Hamill SJ, Sandford R, Thomas RL and Chothia C . 1999 EMBO J 18: 297–305
-
Caelles C, Helmberg A and Karin M . 1994 Nature 370: 220–223
-
Chan TA, Hermeking H, Lengauer C, Kinzler KW and Vogelstein B . 1999 Nature 401: 616–620
-
Chène P . 1998 J Mol Biol 281: 281, 205–209
-
Chène P and Bechter E . 1999 J Mol Biol 286: 1269–1274
-
Cho Y, Gorina S, Jeffrey PD and Pavletich NP . 1994 Science 265: 346–355
-
Clarke J and Fersht AR . 1993 Biochemistry 32: 4322–4329
-
Dalby PA, Oliveberg M and Fersht AR . 1998 J Mol Biol 276: 625–646
-
Di Como CJ and Prives C . 1998 Oncogene 16: 2527–2539
-
Durocher D, Henckel J, Fersht AR and Jackson SP . 1999 Mol Cell 4: 387–394
-
Fahraeus R, Fischer P, Krausz E and Lane DP . 1999 J Pathol 187: 138–146
-
Fersht AR . 1998 Structure and Mechanism in Protein Science A Guide to Enzyme Catalysis and Protein Folding WH Freeman New York
Google Scholar
-
Fersht AR and Serrano L . 1993 Curr Opin Struct Biol 3: 75–83
-
Fourie AM, Hupp TR, Lane DP, Sang BC, Barbosa MS, Sambrook JF and Gething MJ . 1997 J Biol Chem 272: 19471–19479
-
Friedlander P, Legros Y, Soussi T and Prives C . 1996 J Biol Chem 271: 25468–25478
-
Gill SC and von Hippel PH . 1989 Anal Biochem 182: 319–326
-
Gorina S and Pavletich NP . 1996 Science 274: 1001–1005
-
Guillouf C, Rosselli F, Sjin RT, Moustacchi E, Hoffman B and Liebermann DA . 1998 Int J Oncol 13: 107–114
-
Hansen S, Hupp TR and Lane DP . 1996 J Biol Chem 271: 3917–3924
-
Harris CC . 1996 J Natl Cancer Inst 88: 1442–1455
-
Haupt Y, Maya R, Kazaz A and Oren M . 1997a Nature 387: 296–299
-
Haupt Y, Rowan S, Shaulian E, Kazaz A, Vousden K and Oren M . 1997b Leukemia 11: Suppl 3 337–339
-
Haupt Y, Rowan S, Shaulian E, Vousden K and Oren M . 1995 Genes Dev 9: 2170–2183
-
Hernandez-Boussard T, Rodriguez-Tome P, Montesano R and Hainaut P . 1999 Hum Mutat 14: 1–8
-
Hinds PW, Finlay CA, Quartin RS, Baker SJ, Fearon ER, Vogelstein B and Levine AJ . 1990 Cell Growth Differ 1: 571–580
-
Hollstein MC, Sidransky D, Vogelstein B and Harris CC . 1991 Science 253: 49–53
-
Hubbard SJ, Campbell SF and Thornton JM . 1991 J Mol Biol 220: 507–530
-
Hubbard SJ and Thornton JM . 1993 'NACCESS', Computer Program, Department of Biochemistry and Molecular Biology, University College London
-
Hupp TR . 1999 Cell Mol Life Sci 55: 88–95
-
Jayaraman L and Prives C . 1999 Cell Mol Life Sci 55: 76–87
-
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B and Craig RW . 1991 Cancer Res 51: 6304–6311
-
Kirsch DG and Kastan MB . 1998 J Clin Oncol 16: 3158–3168
-
Koradi R, Billeter M and Wüthrich K . 1996 J Mol Graphics 14: 51–55
-
Levine AJ . 1997 Cell 88: 323–331
-
Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE and Jacks T . 1994 Science 266: 807–810
-
Midgley CA and Lane DP . 1997 Oncogene 15: 1179–1189
-
Milner J . 1995 TIBS 20: 49–51
-
Miroux B and Walker JE . 1996 J Mol Biol 260: 289–298
-
Munoz V and Serrano L . 1996 Fold Des 1: R71–R77
-
Myers JK, Pace CN and Scholtz JM . 1995 Pro Sci 4: 2138–2148
-
Nikolova PV, Henckel J, Lane DP and Fersht AR . 1998 Proc Natl Acad Sci USA 95: 14675–14680
-
Ory K, Legros Y, Augguin C and Soussi T . 1994 EMBO J 13: 3496–3504
-
Pace CN . 1986 Methods Enzymol 131: 266–280
-
Pinhashi-Kimhi O, Michalovitz D, Ben-Zeev A and Oren M . 1986 Nature 320: 182–184
-
Polyak K, Xia Y, Zweier JL, Kinzler KW and Vogelstein B . 1997 Nature 389: 300–305
-
Ponchel F and Milner J . 1998 Br J Cancer 77: 1555–1561
-
Radford SE and Dobson CM . 1999 Cell 97: 291–298
-
Randell Brown C, Hong-Brown LQ and Welch WJ . 1997 J Clin Invest 99: 1432–1444
-
Raycroft L, Schmidt JR, Yoas K, Hao MM and Lozano G . 1991 Mol Cell Biol 11: 6067–6074
-
Rolley N, Butcher S and Milner J . 1995 Oncogene 11: 763–770
-
Ryan KM and Vousden KH . 1998 Mol Cell Biol 18: 3692–3698
-
Sauer RT and Lim WA . 1992 Curr Opin Struct Biol 2: 46–51
-
Selivanova G, Ryabchenko L, Jansson E, Iotsova V and Wiman KG . 1999 Mol Cell Biol 19: 3395–3402
-
Serrano L, Kellis Jr JT, Cann P, Matouschek A and Fersht AR . 1992 J Mol Biol 224: 783–804
-
Shaulian E, Zauberman A, Ginsberg D and Oren M . 1992 Mol Cell Biol 12: 5581–5592
-
Soussi T and May P . 1996 J Mol Biol 260: 623–637
-
Thomas PJ, Qu BH and Pedersen PL . 1995 Trends Biochem Sci 20: 456–459
-
Thukral SK, Blain GC, Chang KK and Fields S . 1994 Mol Cell Biol 14: 8315–8321
-
Wacey AI, Cooper DN, Liney D, Hovig E and Krawczak M . 1999 Hum Genet 104: 15–22
-
Walker DR, Bond JP, Tarone RE, Harris CC, Makalowski W, Boguski MS and Greenblatt MS . 1999 Oncogene 18: 211–218
-
Walker KK and Levine AJ . 1996 Proc Natl Acad Sci USA 93: 15335–15340
-
Wang XW, Vermeulen W, Coursen JD, Gibson M, Lupold SE, Forrester K, Xu G, Elmore L, Yeh H, Hoeijmakers JH and Harris CC . 1996 Genes Dev 10: 1219–1232
-
Wiman KG . 1998 Med Oncol 15: 222–228
-
Wong KB, DeDecker BS, Freund SM, Proctor MR, Bycroft M and Fersht AR . 1999 Proc Natl Acad Sci USA 96: 8438–8442
-
Yaffe MB and Cantley LC . 1999 Nature 402: 30–31
-
Zhang W, Guo XY, Hu GY, Liu WB, Shay JW and Deisseroth AB . 1994 EMBO J 13: 2535–2544
Download references
Acknowledgements
We thank Dr Mark Bycroft for his critical reading of the manuscript. We thank Drs Brian DeDecker, Chris Johnson and Kambo Wong for their helpful advice. This work was supported by the CRC of the UK. J Henckel holds a Marie Curie Research Training Grant of the European Commission.
Rights and permissions
About this article
Cite this article
Bullock, A., Henckel, J. & Fersht, A. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245–1256 (2000). https://doi.org/10.1038/sj.onc.1203434
Download citation
-
Received:
-
Revised:
-
Accepted:
-
Published:
-
Issue Date:
-
DOI : https://doi.org/10.1038/sj.onc.1203434
Keywords
- tumour suppressor
- stability
- denaturation
- activity
- protein
Further reading
Can You Draw Back Residual on a Core Pack
Source: https://www.nature.com/articles/1203434